LA MATEMATICA DEGLI ORBITALI MOLECOLARI E DEL LEGAME CHIMICO
Febbraio 7, 2023La teoria del legame di valenza VB e la teoria VSEPR (valence shell electron pair repulsion) sono sufficienti per spiegare la geometria delle molecole e la natura dei legami. Tuttavia per spiegare alcune proprietà quali ad esempio le lunghezze di legame uguali come in SO4-2 si ricorre alla risonanza ma per spiegare il paramagnetismo dell’ossigeno nessuna delle suddette teorie risulta adeguata. Per dare una spiegazione a questo fenomeno e per comprendere meglio altre proprietà quali la natura degli spettri UV ed altre proprietà molecolari ricorriamo alla teoria degli orbitali molecolari.
Il concetto di ORBITALE MOLECOLARE è una naturale estensione del trattamento degli orbitali atomici. Infatti, si assume che due orbitali sovrapponendosi formino una nuvola elettronica che si estende tra i due atomi e quindi gli elettroni appartengono ad ambedue gli atomi e sono soggetti al potenziale di tutti i nuclei ed alla distribuzione della carica degli altri elettroni. Per gli orbitali molecolari (OM) valgono le stesse restrizioni imposte agli orbitali atomici (AO) per esempio un OM può avere al massimo due elettroni e devono avere spin diversi. Anche gli OM devono essere normalizzati perchè la probabilità di trovare l’elettrone nell’OM è il 100 % e ricordando che Ψ² rappresenta la probabilità si deve avere anche per l’OM che chiamiamo sempre Ψ
∫Ψ² dτ =1
e vale anche la condizione di ortogonalità :
∫ΨaΨb dτ = 0
Ciò permette di disegnare fisicamente i contorni delle costanti di probabilità intorno alla molecola e visualizzare le regioni in cui si ha la maggior o minore probabilità di trovare gli elettroni. Possiamo inoltre pensare che ogni OM Ψ sia soluzione dell’equazione di HARTREE- FOCK :
FΨ = ε Ψ
F rappresenta l’amiltoniano di Hartree-Fock
che è simile all’equazione di Schrodinger per un solo elettrone ma che in questo caso tiene conto delle interazioni dei vari elettroni con i diversi nuclei e tra gli stessi elettroni.
Prima di procedere dobbiamo fare alcune considerazioni sugli
ATOMI CON PIU’ ELETTRONI
Hartree ha pensato di approssimare “nel modo migliore possibile” la funzione d’onda che è soluzione dell’equazione di Schrödinger a N elettroni (problema la cui soluzione esatta è in pratica impossibile da ottenere se non con potentissimi computers) con un prodotto di funzioni d’onda a un elettrone che sono gli orbitali atomici. L’esempio più semplice è rappresentato dall’ atomo di elio che ha due elettroni.Infatti l’energia cinetica totale è
T = T1 +T2 =P²x1 +P²y1 +P²z1 + P²x2+P²y2 +P²z2 / 2m (perchè consideriamo il sistema a 3 dimensioni)
e l’energia potenziale è V = – 2e²/r1 -2e²/r2 + e²/r12
pertanto sapendo che l’equazione d’onda tridimensionale è:
d²Ψ/dx2 + d²Ψ/dy2 + d²Ψ/dz2 +8π2m/ h² ( E-V) Ψ = 0
ed essendo che d²/dx2 + d²/dy2 + d²/dz2 si chiama operatore Laplaciano che si indica con ∇ 2 avremo
∇ 2 Ψ + ( 8π2m/ h²) ( E-V) Ψ = 0 l’
Ricordiamoci che Ψ non ha significato fisico e che solo il prodotto tra Ψ e il suo complesso coniugato Ψ* (che non è altro che -iΨ ) cioè ΨΨ* che indicheremo sempre con |Ψ²|.
(Il prodotto ΨΨ* è necessario per rendere reale il numero che ne risulta in quanto Ψ potrebbe assumere qualsiasi valore, anche immaginario).
Per il sistema a due elettroni visto sopra, l’equazione di Hartree-Fock è quindi:
(∇ 2 + ∇ 2 ) Ψ + ( 8π2m/ h²) ( E + 2e²/r1 +2e²/r2 + 2e²/r12 )Ψ
l’ultimo termine V=e²/r12 non è altro che il potenziale di repulsione tra i due elettroni.
Riassumendo diciamo che Hartree-Fock hanno proposto quindi un metodo per ridurre l’equazione di N elettroni ad una somma di equazioni monoelettroniche in grado di trovare una soluzione almeno approssimata all’equazione ad N elettroni.
Essi hanno adoperato un metodo detto SELF CONSISTEND FIELD (SCF) che consisteva nel prendere in considerazione una funzione Ψ che tenesse conto delle interazioni tra elettroni e campo potenziale del nucleo ed il potenziale medio di tutti gli altri elettroni dell’atomo: questo portava ad un’equazione che poteva essere risolta numericamente da cui si poteva ottenere utilizzando un nuovo potenziale un nuovo orbitale Ψ che veniva sostituito nell’equazione per ottenere un altro dato numerico che serviva poi per ottenere una nuovaΨ e si ripeteva il calcolo sino a che non si ottenevano almeno per due volte la stessaΨ.
Le Ψ risultanti da questo calcolo possono essere utilizzate con delle approssimazioni dovute a Slater che ha quindi ottenuto delle Ψ che sono in buon accordo con quelle ottenute con il metodo SCF. Gli orbitali atomici dovuti a Slater ed utilizzati sono:
1s Ne-cr
2s Nr e-cr/2
2px Nxe-cr/2
3s Nr2 e-cr/3
3Px Nxre-cr/3
N è il fattore di normalizzazione ( affinchè ∫ΨΨdτ =1 )
c è la carica nucleare effettiva c=Z -S dove Z= numero atomico ed S= costante di schermo degli altri elettroni )
S=0 per tutti gli elettroni esterni al numero quantico principale considerato
S= 0,35 per ogni elettrone dello stesso numero quantico
S=0,30 è usato per l’elettrone 1s
se l’orbitale considerato è un orbitale s o p allora S=0,85 per ogni elettrone che si trova nello strato vicino mentre S=1,00 per ogni elettrone ulteriore nello stesso strato.
In definitiva si può calcolare facilmente che la carica nucleare effettiva Z
per il carbonio è
C1s =5,70 C2p= 3,25
per l’azoto N è
N2p = 3,90
per l’ossigeno O è
O2p= 4,55
Adesso sappiamo quali siano le espressioni matematiche di Ψ1s Ψ2s ecc. quindi vediamo come esse vengono utilizzate nel calcolo degli orbitali molecolari.
Prendiamo come esempio la molecola di idrogeno H2 : essa contiene 2 elettroni ognuno proveniente da un atomo H e che contribuiscono a formare la molecola . Poichè non possiamo distinguere a quali atomi gli elettroni appartengano nella molecola, facciamo una semplificazione razionale: supponiamo che quando un elettrone si trova vicino all’atomo A la funzione d’onda sia Ψa cioè la funzione d’onda che descrive l’elettrone sull’atomo A e che quando un elettrone si trova vicino all’atomo B la funzione d’onda sia Ψb.
Possiamo quindi ragionevolmente pensare che la combinazione lineare degli orbitali Ψa e Ψb possano rappresentare l’orbitale molecolare Ψm o meglio semplicemente Ψ. Pertanto :
Ψ = Ca Ψa + Cb Ψb
Ψ = Ca Ψa – Cb Ψb
in cui Ca e Cb rappresentano l’entità del contributo dei due orbitali atomici nella formazione della molecola. Ovviamente se vi sono più atomi allora l’OM sarà:
Ψ = Ca Ψa + Cb Ψb + Cc Ψc + ….CnΨn
o meglio

dove con χi indichiamo gli orbitali atomici al posto di Ψ.
Il metodo descritto sopra è la cosiddetta approssimazione LCAO (linear combination of atomic orbitals).
Le funzioni d’onda, utilizzabili con questo metodo, devono avere più o meno la stessa grandezza di energia.
Per calcolare l’energia dell’OM dato sopra occorre sostituire la funzione Ψ molecolare nell’equazione d’onda
H Ψ = EΨ
moltiplichiamo ambedue i termini per Ψ* ed integriamo. Avremo:
∫ΨHΨ*δτ = ∫EΨΨ*δτ=E∫ΨΨ*δτ*
da cui
E= ∫ΨHΨ*δτ /∫ΨΨ*δτ
e sostituendo a Ψ la funzione OM che riscriviamo per comodità sostituendo a a Ca e b a Cb
Ψ = a Ψa + b Ψb
si ha
E= ∫ a Ψa + b Ψb H a Ψa + b Ψb dt / ∫ (a Ψa + b Ψb ) (a Ψa + b Ψb) dt
E= ∫( a Ψa + b Ψb Ha Ψa + b Ψb) dt / ∫ (a Ψa + b Ψb ) (a Ψa + b Ψb) dt
e sviluppando si ha
∫aψ*aHaψadt+∫aψ*aHbψbdt+∫bψ*bHaψadt+∫bψ*bHbψbdt/∫aψ*aaψadt+∫aψ*abψbdt+∫bψ*baψadt+∫bψ*bbψbdt
a2∫ψ*aHψadt+ab∫ψ*aHψbdt+ba∫ψ*bHψadt+b2∫ψ*bHψbdt/a2∫ψ*aψadt+ab∫ψ*aψbdt+ba∫ψ*bψadt+b2∫ψ*bψbdt
ponendo
Haa = ∫ ΨaHΨadt Hab= ∫ ΨaHΨbdt
Hba = ∫ ΨbHΨadt Hbb = ∫ ΨbHΨbdt
Saa =∫ ΨaΨadt Sab = ∫ ΨaΨbdt
Sba = ∫ ΨbΨadt Sbb =∫ ΨaHΨadt
possiamo scrivere
E = a² Haa + ab Hab + ba Hba +b² Hbb / a² Saa + ab Sab + ba Sba + b² Sbb
I valori di a e b che rendono minima l’energia (più bassa è l’energia del legame più stabile è la molecola) si ottengono derivando l’equazione scritta sopra prima rispetto ad a poi rispetto a b ed eguagliando a zero
δE/δa =0
δE/δb =0
ricordando che la derivata di una frazione è data da
d/da = [(derivata del numeratore rispetto ad a) x (il denominatore non derivato) – (il numeratore non derivato) x (la derivata del denominatore)] / (denominatore )²
a (Haa-SaaE) + b (Hab -SabE) =0
a(Hba-SbaE) + b(Hbb– SbbE) =0
Possiamo riscrivere più semplicemente le equazioni nel seguente modo:
a∫Ψa (H-E)Ψa dt +b ∫Ψa (H-E)Ψb dt =0
a∫Ψb (H-E)Ψa dt + b ∫Ψb (H-E)Ψb dt =0
questo risulta in due equazioni:
a (Haa-E) + b (Hab -E Sab) =0
a(Hba – E Sba) + b( Hbb – E ) =0
noi siamo interessati a soluzioni in cui a e b devono essere diversi da zero, quindi è necessario che il determinante
| Haa -E Hab -ESab |
| Hba-ESba Hbb – E | =0
(sia uguale a zero).
questo determinante viene chiamato DETERMINANTE SECOLARE e una volta risolto
(Haa-E ) (Hbb-E) – (Hab-ESab)(Hba-ESba) =0
se poniamo adesso
Haa= Hbb= α
Hab=Hba = β è chiamato integrale di scambio e rappresenta la parte di energia dovuta ad entrambi i nuclei sugli elettroni
Saa =Sbb = 1
Sab= Sba=0 è chiamato integrale di sovrapposizione
il determinante scritto sopra diviene :
α-E β
β α-E
e dividendo tutto per β
|α-E/β 1 |
| 1 α-E/β | =0 e ponendo α-E/β = X
risolvendo il determinante si ha
X² -1 =0 cioè X=1 X=-1 pertanto α – E1/β = -1 da cui E1 = α + β
Pertanto l’energia associata a Ψ1 è E = α + β in questo caso l’orbitale è chiamato “bonding“
l’energia associata a Ψ2 è E= α – β in questo caso l’orbitale è detto “antibonding”
possiamo adesso indicare in un diagramma le energie degli orbitali al variare della distanza nucleare
Nel caso dell’idrogeno si ha il seguente diagramma di energia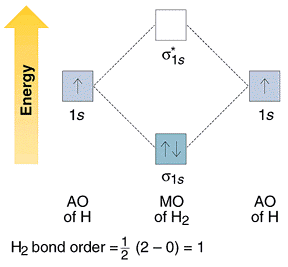
come si vede, gli elettroni provenienti dai due atomi di idrogeno adesso occupano l’orbitale a più bassa energia (orbitale σ ) con le stesse regole utilizzate per gli orbitali atomici (ad es. un OM può essere occupato solo da 2 elettroni e con spin diversi).
Per ottenere,gli orbitali molecolari con il metodo LCAO si devono osservare le seguenti regole:
1- si possono combinare solo gli orbitali atomici che posseggono energia non troppo diversa tra loro
2- esiste un criterio di massima sovrapposizione e se l’integrale di sovrapposizione è nullo allora anche l’integrale di scambio è nullo. Questo implica che solo gli orbitali più esterni possono venir combinati
3- si possono combinare solo gli orbitali atomici che possiedono la stessa simmetria rispetto all’asse di legame
ESEMPIO DI CALCOLO PER L’ETILENE
La molecola di etilene H2C=CH2 può essere sinteticamente rappresentata nel modo seguente in cui si indica con 1 l’orbitale Pz dell’atomo di C1 e con 2 l’orbitale Pz del C2. Abbiamo pertanto gli orbital ΨP1 e Ψ P2. (che indicheremo con P1 e P2 )che contribuiscono a formare l’orbitale molecolare ΨM.
Possiamo scrivere allora ΨM = a1 P1 + a2 P2
Sostituendo questa funzione nell’equazione di Schrodinger HΨ = EΨ si ha:
H(a1P1 +a2P2) = E(a1P1+a2P2) e moltiplicando per ΨM si ha:
(a1P1 +a2P2)H(a1P1 +a2P2) = E(a1P1+a2P2) a1P1 +a2P2)
da cui integrando si ha : ∫(a1P1 +a2P2)H(a1P1 +a2P2) = E1P1+a2P2) a1P1 +a2P2)
cioè
E=∫(a1P1+a2P2)H(a1P1+a2P2)/∫(a1P1+a2P2)(a1P1+a2P2)
e sviluppando
E= a12∫P1P1dt+a1a2∫P1P2dt+a2a1∫P2P1dt+a22∫P2HP2dt /∫P1P1dt+a1a2∫P1P2dt+a2a1∫P2P1dt+a22∫p2Hp2dt
derivando rispetto ad a ed a1ed a2 ed uguagliando a zero
δE/δa1 =0 e δE/δa2 =0
si ha ricordando che la derivata di un quoziente è
δu/δv =vδu- uδv / v²
δE/δa1=0= (2a1H11+a2H12+a2H21+0)(a12S11+a1a2S12+a2a1S21+a22S22)−((a12H11+a1a2H12+a2a1H21+a22H22)(2a1S11+a2S12+a2S21+0) /(a12S11+a1a2S12+a2a1S21+a22S22)(a12S11+a1a2S12+a2a1S21+a22S22)
dove il termine frazionario dopo il segno meno
(a12H11+a1a2H12+a2a1H21+a22H22)(a12S11+q1a2S12+a2a1S21+a22S22) =E
possiamo scrivere quindi:
δE/δa1=0=(2a1H11+a2H12+a2H21)−(2a1S11+a2S12+a2S21)E
(analogo calcolo viene effettuato per a2 )
δE/δa1=0=2(a1H1−a1S11E)+a2(H12−S12E)+a2H21(−S21E)
2a1(H11-S11E) + 2a2(H12-S12E)=0
e dividendo per 2
a1(H11-S11E) + a2(H12-S12E)=0
indicando l’integrale H11=α e l’integrale H12=H21 =β si ha il determinante che risolto fornisce
(α-E) (α-E) – β² =0 da cui si ottengono le due radici di E
E1=α+β ed E2=α-β
ed essendo β un valore negativo allora E1<E2
I due elettroni π dell’etilene occupano il livello di energia più basso cioè E1 per cui l’energia π della molecola è
E = 2 (α+β)
Se i due elettroni della molecola vengono eccitati, uno di essi occupa il livello di energia E2 per cui l’energia π della molecola diviene
E1= (α+β) + (α-β) = 2α
e l’energia della transizione diviene E2-E1= -2β
CALCOLO DELLA FUNZIONE Ψ CHE CORRISPONDE ALL’ENERGIA E1
Sappiamo che la funzione Ψ ha la seguente forma : Ψ = c11χ1 + c12χ2
ed il sistema associato al determinante è :
c11(α -E1) + c12 β =0
c11β + c12(α -E1)=0
e si riduce ad una sola equazione essendo il determinante corrispondente nullo.
c11(α -E1) +c12β =0
ma essendo E1= α+β si ha -c11β +c12β =0
Essendo l’integrale β diverso da zero allora deve essere c11=c12
la funzione cercata ha quindi la forma Ψ1 = c11(χ1 + χ2)
Il valore di C11 viene calcolato dalla condizione di normalizzazione c112 + c122 =1
essendo c11=c12 allora C11=C12= ± 1 / √2 pertanto la funzione d’onda corrispondente all’energia E1 della molecola di etilene è Ψ1 = 1 / √2 (χ1 + χ2).
CALCOLO DELLA CARICA π NELL’ETILENE
Nello stato fondamentale le cariche elettroniche π sono
q1= 2C112 =1
q2=2C122 =1
nel primo stato eccitato
q1′ = c112 + c122 =1
q2′ = c122 + c222 =1
CALCOLO DEGLI INDICI DI LEGAME
nello stato fondamentale L12= 2C11 C12 =1
nel primo stato eccitato L’12 =c11c12 +c21c22 =0
